Gene Checker
Overview
 Medicover's testing kits provide a wide range of highly reliable and easy-to-implement genetic testing solutions, offering accurate, rapid, and cost-effective detection of genetic mutations.
Medicover's testing kits provide a wide range of highly reliable and easy-to-implement genetic testing solutions, offering accurate, rapid, and cost-effective detection of genetic mutations.
The testing kits consist of NGS-based genetic panel testing kits, including reagents for universal library preparation and target capture enrichment workflows covering multiple fields. By running multiple assays in the same sequencing run, assays from all testing kits can be integrated into a unified workflow. Medicover's testing kits offer the uniqueness of performing multi-field genetic testing simultaneously in one sequencing run, ensuring accurate and rapid detection of genetic mutations while enhancing cost and operational efficiency in labs of all throughput capabilities.
Manufactured under strict quality control processes (ISO 13485:2016 and ISO 9001:2015), the testing kits are library preparation and enrichment kits with guaranteed highest quality, intended for the identification of genetic mutations associated with diseases.
Running Multiple Assays in One Run
- Highly assured assays
- Comprehensive genetic content providing clinically meaningful information
- User-friendly and highly reliable workflows and protocols
- Integration of assays to provide high operational efficiency
- Comprehensive system including software solutions and result reporting
Specifications and Features
- Common protocol for all assays
- Simplified and streamlined workflow
- Common sample requirements
- Compatibility with automation
- Compatibility with laboratories of all throughputs
- Sample types: Oral swab (except for PGT: polar body or blastocyst biopsy)
- Uniformity of average coverage ≥ 20x: >97%
- Proprietary bioinformatics pipeline
- Proprietary dual indexing compatible with all testing kits
Assay Menu
Medicover's testing kits provide a unique and efficient solution for conducting multi-field genetic testing within your organization, catering to labs of all throughputs. By combining various assays in one sequencing run, it's possible to shorten turnaround times and reduce operational costs while ensuring high-quality results. Our comprehensive testing approach is ideal for labs looking to offer a wide range of reliable genetic tests to healthcare professionals and patients.
The testing kits cover the following areas:
Oncology
Postnatal Testing
Neonatal
Reproductive Health
Preimplantation Genetic Testing (PGT)
Target Diseases for Genetic Testing
Carrier Core Kit
| Disorder | Gene |
|---|---|
| Alpha Thalassemia | HBA1, HBA2 |
| Beta Thalassemia | HBB |
| Bloom Syndrome | BLM |
| Canavan Disease | ASPA |
| Cystic Fibrosis | CFTR |
| Duchenne Muscular Dystrophy, X-linked | DMD |
| Family Autonomic Dysfunction | ELP1 |
| Fanconi Anemia, Type C | FANCC |
| Galactosemia | GALT |
| Gaucher Disease | GBA |
| Medium Chain Acyl-CoA Dehydrogenase Deficiency | ACADM |
| Mucolipidosis, Type IV | MCOLN1 |
| Niemann-Pick Disease, Type A/B | SMPD1 |
| Phenylalanine Hydroxylase Deficiency | PAH |
| Sickle Cell Anemia | HBB |
| Smith-Lemli-Opitz Syndrome | DHCR7 |
| Tay-Sachs Disease | HEXA |
| Non-Syndromic Hearing Loss GJB2 Related / DFNB1 Related | GJB2, GJB6 |
Career Comprehensive Kit
| Disorder | Gene |
|---|---|
| 3-Hydroxy-3-Methylglutaryl-CoA Lyase Deficiency | HMGCL |
| 3-Methylcrotonyl-CoA Carboxylase Deficiency 1 | MCCC1 |
| 3-Methylcrotonyl-CoA Carboxylase Deficiency 2 | MCCC2 |
| 3-Methylglutaconic Aciduria Type 3 [Costeff Syndrome] | OPA3 |
| 3-Phosphoglycerate Dehydrogenase Deficiency | PHGDH |
| 6-Pyruvoyl-Tetrahydropterin Synthase (PTPS) Deficiency | PTS |
| Abetalipoproteinemia | MTTP |
| Sulfate Transporter-Related Osteochondrodysplasia | SLC26A2 |
| Achromatopsia (CNGB3-Related) | CNGB3 |
| Acute Infantile Liver Failure (TRMU-Related) | TRMU |
| Acyl-CoA Oxidase I Deficiency | ACOX1 |
| X-Linked Adrenoleukodystrophy | ABCD1 |
| Aicardi-Goutières Syndrome | SAMHD1 |
| Alpha Thalassemia | HBA1, HBA2 |
| Alport Syndrome (COL4A3-Related) | COL4A3 |
| Alport Syndrome, X-Linked | COL4A5 |
| Alström Syndrome | ALMS1 |
| Andersen Syndrome | SLC12A6 |
| Argininosuccinate Lyase Deficiency | ASL |
| Aromatase Deficiency | CYP19A1 |
| Articulation Contracture, Intellectual Disability, Seizures | SLC35A3 |
| Asparagine Synthetase Deficiency | ASNS |
| Aspartylglucosaminuria | AGA |
| Ataxia with Vitamin E Deficiency | TTPA |
| Ataxia-Telangiectasia | ATM |
| Autoimmune Polyendocrine Syndrome, Type 1 | AIRE |
| Autosomal Recessive Polycystic Kidney Disease | PKHD1 |
| Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay | SACS |
| Bardet-Biedl Syndrome (BBS1-Related) | BBS1 |
| Bardet-Biedl Syndrome, Type 12 (BBS12-Related) | BBS12 |
| Bare Lymphocyte Syndrome (CIITA-Related) | CIITA |
| Bartter Syndrome (BSND-Related) | BSND |
| Batten Disease (CLN3-Related) | CLN3 |
| Beta Thalassemia | HBB |
| Biotinidase Deficiency | BTD |
| Bloom Syndrome | BLM |
| Canavan Disease | ASPA |
| Carnitine Palmitoyltransferase IA Deficiency | CPT1A |
| Carnitine Palmitoyltransferase II Deficiency | CPT2 |
| Coarctation of the Aorta Syndrome | RAB23 |
| Cartilage-Hair Hypoplasia | RMRP |
| Cerebrotendinous Xanthomatosis | CYP27A1 |
| Chorea-Acanthocytosis | VPS13A |
| Choroideremia, X-Linked | CHM |
| Chronic Granulomatous Disease, X-Linked | CYBB |
| Citrullinemia | SLC25A13 |
| Citrullinemia Type I | ASS1 |
| Combined Malonic and Methylmalonic Acidemia | ACSF3 |
| Combined Oxidative Phosphorylation Deficiency 1 | GFM1 |
| Combined Oxidative Phosphorylation Deficiency 3 | TSFM |
| Combined Pituitary Hormone Deficiency 2 | PROP1 |
| Congenital Disorder of Glycosylation, Type 1A (PMM2-Related) | PMM2 |
| Congenital Disorder of Glycosylation, Type 1B | MPI |
| Congenital Disorder of Glycosylation Type 1C | ALG6 |
| Congenital Nephrotic Syndrome | NPHS1 |
| Congenital Insensitivity to Pain with Anhidrosis | NTRK1 |
| Congenital Myasthenic Syndrome (CHRNE-Related) | CHRNE |
| Congenital Myasthenic Syndrome (RAPSN-Related) | RAPSN |
| Congenital Neutropenia (HAX1-Related) | HAX1 |
| Congenital Neutropenia (VPS45-Related) | VPS45 |
| Corneal Dystrophy and Perceptive Deafness | SLC4A11 |
| Corticosterone Methyloxidase Deficiency | CYP11B2 |
| CRB1-Related Retinal Dystrophy | CRB1 |
| Creatine Transporter Deficiency [Cerebral Creatine Deficiency Syndrome 1] X-Linked | SLC6A8 |
| Crigler-Najjar Syndrome, Types I/II | UGT1A1 |
| Cystic Fibrosis | CFTR |
| Cystinosis | CTNS |
| D-Bifunctional Protein Deficiency | HSD17B4 |
| Deafness, Autosomal Recessive 77 | LOXHD1 |
| Duchenne Muscular Dystrophy, X-Linked | DMD |
| Dystrophic Epidermolysis Bullosa (COL7A1-Related) | COL7A1 |
| Ehlers-Danlos Syndrome, Type VIIC | ADAMTS2 |
| Emery-Dreifuss Muscular Dystrophy 1, X-Linked | EMD |
| S-Con Syndrome Enhancer | NR2E3 |
| Ethylmalonic Encephalopathy | ETHE1 |
| Fabry Disease, X-Linked | GLA |
| Factor IX Deficiency, X-Linked | F9 |
| Factor V Leiden Thrombophilia | F5 |
| Factor XI Deficiency | F11 |
| Familial Dysautonomia | ELP1 |
| Familial Hypercholesterolemia (LDLR-Related) | LDLR |
| Familial Mediterranean Fever | MEFV |
| Nephrogenic Diabetes Insipidus (AQP2-Related) | AQP2 |
| Fanconi Anemia, Type G | FANCG |
| Fanconi Anemia, Type C | FANCC |
| Galactokinase Deficiency (Galactosemia, Type II) | GALK1 |
| Galactosemia | GALT |
| Gaucher Disease | GBA |
| Glutaric Acidemia Type 1 | GCDH |
| Glutaric Acidemia Type 2A | ETFA |
| Glycine Encephalopathy (GLDC-Related) | GLDC |
| Glycine Encephalopathy (AMT-Related) | AMT |
| Glycogen Storage Disease Type 1A | G6PC |
| Glycogen Storage Disease Type 1 | SLC37A4 |
| Glycogen Storage Disease, Type 2 (Pompe Disease) | GAA |
| Glycogen Storage Disease, Type 3 | AGL |
| Glycogen Storage Disease Type 4 | GBE1 |
| Glycogen Storage Disease, Type 5 (McArdle Disease) | PYGM |
| Glycogen Storage Disease Type 7 | PFKM |
| Griscelli Syndrome | BCS1L |
| Hemochromatosis Type 2A | HJV |
| Hemochromatosis, Type 3 (TFR2-Related) | TFR2 |
| Hereditary Fructose Intolerance | ALDOB |
| Hermansky-Pudlak Syndrome Type 1 (HPS1-Related) | HPS1 |
| Hermansky-Pudlak Syndrome Type 3 (HPS3-Related) | HPS3 |
| Holocarboxylase Synthetase Deficiency | HLCS |
| Homocystinuria (CBS-Related) | CBS |
| Homocystinuria, cblE Type | MTRR |
| Hydrolethalus Syndrome | HYLS1 |
| Hypohidrotic Ectodermal Dysplasia, X-Linked | EDA |
| Hypophosphatasia (ALPL-Related) | ALPL |
| Inclusion Body Myopathy 2 | GNE |
| Isovaleryl-CoA Dehydrogenase Deficiency | IVD |
| Joubert Syndrome 2 | TMEM216 |
| Joubert Syndrome 2 | TMEM216 |
| Junctional Epidermolysis Bullosa, Herlitz Type | LAMC2 |
| Juvenile Retinoschisis, X-Linked | RS1 |
| Krabbe Disease | GALC |
| Lamellar Ichthyosis, Type 1 | TGM1 |
| Leber Congenital Amaurosis, CEP290 Type | CEP290 |
| Leigh Syndrome, French-Canadian Type | LRPPRC |
| Leukodystrophy with Vanishing White Matter | EIF2B5 |
| Leydig Cell Hypoplasia [Luteinizing Hormone Resistance] | LHCGR |
| Limb-Girdle Muscular Dystrophy, Type 2A | CAPN3 |
| Limb-Girdle Muscular Dystrophy, Type 2B | DYSF |
| Limb-Girdle Muscular Dystrophy, Type 2C | SGCG |
| Limb-Girdle Muscular Dystrophy, Type 2D | SGCA |
| Limb-Girdle Muscular Dystrophy, Type 2E | SGCB |
| Lipoamide Dehydrogenase Deficiency [Maple Syrup Urine Disease, Type 3] | DLD |
| Lipoid Adrenal Hyperplasia | STAR |
| Lipoprotein Lipase Deficiency | LPL |
| Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency | HADHA |
| Lysinuric Protein Intolerance | SLC7A7 |
| Maple Syrup Urine Disease, Type 1B | BCKDHB |
| Meckel-Gruber Syndrome Type 1 | MKS1 |
| Medium-Chain Acyl-CoA Dehydrogenase Deficiency | ACADM |
| Megalencephalic Leukoencephalopathy with Subcortical Cysts | MLC1 |
| Metachromatic Leukodystrophy (ARSA-related) | ARSA |
| Metachromatic Leukodystrophy (PSAP-related) | PSAP |
| Methylmalonic Aciduria (MMAA-related) | MMAA |
| Methylmalonic Aciduria (MMAB-related) | MMAB |
| Methylmalonic Aciduria and Homocystinuria, cblC Type | MMACHC |
| Methylmalonic Aciduria and Homocystinuria, cblD Type | MMADHC |
| Methylmalonic Aciduria, Mut(0) Type | MMUT |
| Microphthalmia/Anophthalmia (VSX2-related) | VSX2 |
| Mitochondrial Complex I Deficiency (ACAD9-related) | ACAD9 |
| Mitochondrial Complex I Deficiency (NDUFAF5-related) | NDUFAF5 |
| Mitochondrial Complex I Deficiency (NDUFS6-related) | NDUFS6 |
| Mitochondrial Myopathy and Sideroblastic Anemia (MLASA1) | PUS1 |
| Mucolipidosis II/III | GNPTAB |
| Mucolipidosis III gamma | GNPTG |
| Mucolipidosis, Type IV | MCOLN1 |
| Mucopolysaccharidosis II [Hunter Syndrome], X-Linked | IDS |
| Mucopolysaccharidosis IIIB [Sanfilippo B] | NAGLU |
| Mucopolysaccharidosis IIIC [Sanfilippo C] | HGSNAT |
| Mucopolysaccharidosis IIID [Sanfilippo D] | GNS |
| Mucopolysaccharidosis, Type IX | HYAL1 |
| Multiple Sulfatase Deficiency | SUMF1 |
| Myoneurogastrointestinal Encephalopathy (MNGIE) | TYMP |
| Myotubular Myopathy, X-Linked | MTM1 |
| N-Acetylglutamate Synthase Deficiency | NAGS |
| Navajo Neurohepatopathy [MPV17-related Hepatocerebral Mitochondrial DNA Depletion Syndrome] | MPV17 |
| Neuronal Ceroid Lipofuscinosis (TPP1-related) | TPP1 |
| Neuronal Ceroid Lipofuscinosis (MFSD8-related) | MFSD8 |
| Neuronal Ceroid Lipofuscinosis (CLN5-related) | CLN5 |
| Neuronal Ceroid Lipofuscinosis (CLN6-related) | CLN6 |
| Neuronal Ceroid Lipofuscinosis (CLN8-related) | CLN8 |
| Neuronal Ceroid Lipofuscinosis (PPT1-related) | PPT1 |
| Niemann-Pick Disease, Type A/B | SMPD1 |
| Niemann-Pick Disease, C1/D Type | NPC1 |
| Niemann-Pick Disease, C2 Type | NPC2 |
| Nijmegen Breakage Syndrome | NBN |
| Non-Syndromic Hearing Loss (GJB2, GJB6-related) | GJB2, GJB6 |
| Odonto-Onycho-Dermal Dysplasia/Schopf-SchulzPassarge Syndrome | WNT10A |
| Omen Syndrome, RAG2-related | RAG2 |
| Ornithine Aminotransferase Deficiency | OAT |
| Ornithine Transcarbamylase Deficiency | OTC |
| Ornithine Translocase Deficiency [Hyperornithinemia-Hyperammonemia-Homocitrullinuria (HHH) Syndrome] | SLC25A15 |
| Pendred Syndrome | SLC26A4 |
| Peroxisome Biogenesis Disorder Zellweger Syndrome Spectrum (PEX1-related) | PEX1 |
| Peroxisome Biogenesis Disorder Zellweger Syndrome Spectrum (PEX2-related) | PEX2 |
| Phenylalanine Hydroxylase Deficiency | PAH |
| Pituitary Hormone Deficiency, Combined, Type 3 | LHX3 |
| Pontocerebellar Hypoplasia, RARS2-related | RARS2 |
| Pontocerebellar Hypoplasia, Type 1A | VRK1 |
| Pontocerebellar Hypoplasia, Type 2D | SEPSECS |
| Pontocerebellar Hypoplasia, Type 2E | VPS53 |
| Primary Ciliary Dyskinesia, DNAH5-related | DNAH5 |
| Primary Ciliary Dyskinesia, DNAI1-related | DNAI1 |
| Primary Ciliary Dyskinesia, DNAI2-related | DNAI2 |
| Primary Hyperoxaluria, Type 1 | AGXT |
| Primary Hyperoxaluria, Type 2 | GRHPR |
| Primary Hyperoxaluria, Type 3 | HOGA1 |
| Porokeratosis | CTSK |
| Pyruvate Dehydrogenase Deficiency (PDHB-related) | PDHB |
| Pyruvate Dehydrogenase Deficiency, X-Linked | PDHA1 |
| Renal Tubular Acidosis and Deafness (ATP6V1B11-related) | ATP6V1B1 |
| Retinal Dystrophy (RLBP1-related) [Bosnia Retinal Dystrophy] | RLBP1 |
| Retinitis Pigmentosa 59 (DHDDS-related) | DHDDS |
| Retinitis Pigmentosa 25 (EYS-related) | EYS |
| Retinitis Pigmentosa 26 | CERKL |
| Retinitis Pigmentosa 28 | FAM161A |
| X-Linked Retinitis Pigmentosa | RPGR |
| Rhizomeric Chondrodysplasia Punctata Type 1 | PEX7 |
| Rhizomeric Chondrodysplasia Punctata Type 3 | AGPS |
| Roberts Syndrome | ESCO2 |
| Salla Disease | SLC17A5 |
| Sandhoff Disease | HEXB |
| Schimke Immunoosseous Dysplasia | SMARCAL1 |
| Segawa Syndrome (TH-related) | TH |
| Severe Combined Immunodeficiency, Ataxia Type | DCLRE1C |
| Severe Combined Immunodeficiency, X-Linked | IL2RG |
| Sickle Cell Anemia | HBB |
| Chédiak-Higashi Syndrome | ALDH3A2 |
| Smith-Lemli-Opitz Syndrome | DHCR7 |
| Steroid-Resistant Nephrotic Syndrome | NPHS2 |
| Stuve-Wiedemann Syndrome | LIFR |
| Tay-Sachs Disease | HEXA |
| Type 1 Tyrosinemia | FAH |
| Usher Syndrome Type 1C | USH1C |
| Usher Syndrome Type 1F | PCDH15 |
| Usher Syndrome Type 2A | USH2A |
| Usher Syndrome Type 3 | CLRN1 |
| Wilson Disease | ATP7B |
| Wolman Disease | LIPA |
| Zellweger Spectrum Disorders (PEX6-related) | PEX6 |
| Zellweger Spectrum Disorders (PEX10-related) | PEX10 |
Hereditary Cancer Kit
| Disorder | Gene |
|---|---|
| Ataxia-Telangiectasia Syndrome | ATM |
| BAP1 Tumor Predisposition Syndrome | BAP1 |
| Hereditary Nonpolyposis Colorectal Cancer | EPCAM, MSH2, MSH6, MLH1, PMS2 |
| DICER1 Syndrome | DICER1 |
| Familial Adenomatous Polyposis / Attenuated Familial Adenomatous Polyposis | APC |
| Fanconi Anemia | FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, SLX4, ERCC4, BRCA1, BRCA2, BRIP1, PALB2, RAD51C |
| Hereditary Breast and Ovarian Cancer Syndrome | BRCA1, BRCA2 |
| Hereditary Diffuse Gastric Cancer | CDH1 |
| Hereditary Mixed Polyposis Syndrome | GREM1 |
| Hereditary Melanoma-Pancreatic Cancer Syndrome | CDKN2A, CDK4 |
| Hereditary Paraganglioma-Pheochromocytoma Syndrome | SDHD, SDHAF2, SDHC, SDHB |
| Juvenile Polyposis Syndrome | SMAD4, BMPR1A |
| Li-Fraumeni Syndrome | TP53 |
| Li-Fraumeni Syndrome 2 | CHEK2 |
| Lynch Syndrome | EPCAM, MLH1, MSH2, MSH6, PMS2 |
| Multiple Endocrine Neoplasia Type 1 | MEN1 |
| Multiple Endocrine Neoplasia Type 2 | RET |
| MUTYH-Associated Polyposis Syndrome | MUTYH |
| Poitz-Jeghers Syndrome | STK11 |
| Polymerase Proofreading-related Syndrome | POLD1, POLE |
| PTEN Hamartoma Syndrome | PTEN |
| Retinoblastoma | RB1 |
| Von Hippel-Lindau Syndrome | VHL |
| Xeroderma Pigmentosum Syndrome | DDB2, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, POLH, XPA, XPC |
Cancers Associated with This Panel:
Breast, Ovarian, Endometrial or Associated Cancers, Colorectal, Blood, Gastric (stomach), Prostate, Pancreatic, Renal (kidney), Skin, Paraganglioma or Pheochromocytoma, Familial Melanoma Skin, Parathyroid, Medullary thyroid
Infertility Kit
Female Infertility
| Disorder | Gene |
|---|---|
| Primary Ovarian Insufficiency | BMP15 |
| CYP19A1 | |
| FOXL2 | |
| GALT | |
| GNAS | |
| GNRHR | |
| KISS1R | |
| NOBOX | |
| STAG3 | |
| ZP1 | |
| EIF2B2 | |
| EIF2B3 | |
| FIGLA | |
| POF1B | |
| POLG | |
| PSMC3IP | |
| WT1 | |
| FSHB | |
| NR5A1 | |
| LHB | |
| LHCGR | |
| FSHR | |
| CYP17A1 | |
| GDF9 | |
| AIRE | |
| Polycystic Ovary Syndrome | LHCGR |
| FSHR | |
| CYP17A1 | |
| GDF9 | |
| GNRH1 | |
| INS | |
| INSR | |
| IRS1 | |
| IRS2 | |
| KISS1 | |
| TACR3 | |
| CAPN10 | |
| CYP11A1 | |
| DENND1A | |
| THADA | |
| Hypogonadotropic Hypogonadism | GNRHR |
| KISS1R | |
| FSHB | |
| LHB | |
| GNRH1 | |
| KISS1 | |
| TACR3 | |
| ANOS1 | |
| CHD7 | |
| DUSP6 | |
| FEZF1 | |
| FGF17 | |
| FGF8 | |
| FGFR1 | |
| FLRT3 | |
| HESX1 | |
| HS6ST1 | |
| IL17RD | |
| NSMF | |
| PROK2 | |
| PROKR2 | |
| SEMA3A | |
| SPRY4 | |
| TAC3 | |
| WDR11 | |
| Ovarian Hyperstimulation Syndrome | FSHR |
Male Infertility
| Disorder | Gene |
|---|---|
| Spermatogenic Failure (Azoospermia, Oligospermia, etc.) | ANOS1(KAL1) |
| AR | |
| AURKC | |
| CATSPER1 | |
| CFTR | |
| CHD7 | |
| DAZL | |
| DDX25 | |
| FGF8 | |
| FSHB | |
| FSHR | |
| NR5A1 | |
| PRM1 | |
| USP26 | |
| USP9Y | |
| Disorders of Sexual Development | AR |
| LHCGR | |
| NR5A1 | |
| SRY | |
| SRD5A1 | |
| Congenital Bilateral Absence of the Vas Deferens (CBAVD) | CFTR |
| Hypogonadotropic Hypogonadism (HH) | ANOS1(KAL1) |
| CHD7 | |
| DUSP6 | |
| FEZF1 | |
| FGF17 | |
| FGF8 | |
| FLRT3 | |
| FSHB | |
| GNRH1 | |
| GNRHR | |
| HESX1 | |
| HS6ST1 | |
| IL17RD | |
| KISS1 | |
| KISS1R | |
| LHB | |
| NR5A1 | |
| NSMF | |
| PROK2 | |
| PROKR2 | |
| SEMA3A | |
| SPRY4 | |
| TAC3 | |
| TACR3 | |
| WDR11 | |
| FGFR1 | |
| LHCGR |
Newborn Screening Kit
| Endocrine Disorders | Genes |
|---|---|
| Congenital Adrenal Hyperplasia | CYP11B1, CYP17A1, HSD3B2, POR, STAR |
| Congenital Hypothyroidism | PAX8, THRA, SLC5A5, TG, TPO, TSHB, TSHR |
| Pendred Syndrome | SLC26A4 |
| Hemoglobin | Gene |
|---|---|
| β-Thalassemia | HBB |
| S, β-Thalassemia (Sickle Cell β-Thalassemia) | HBB |
| S,C Disease (Sickle Cell Disease) | HBB |
| S,S Disease (Sickle Cell Disease, Sickle Cell Anemia) | HBB |
| Hearing Loss | Gene |
|---|---|
| Non-syndromic Hearing Loss | CDH23, MYO15A, OTOF, TMIE, TMPRSS3, TPRN, TRIOBP, GJB2, GJB6, TECTA |
| Syndromic Hearing Loss | |
| Jervell and Lange-Nielsen Syndrome | KCNE1, KCNQ1 |
| Pendred Syndrome | SLC26A4 |
| Waardenburg Syndrome | SOX10 |
| Usher Syndrome 1C | USH1C |
| Usher Syndrome 1G | USH1G |
| Usher Syndrome 2A | USH2A |
| Usher Syndrome IID | WHRN |
| Waardenburg Syndrome | PAX3 |
| Metabolic | Gene |
|---|---|
| 2,4-Dienoyl-CoA Reductase Deficiency | NADK2 |
| 2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency | HSD17B10 |
| 2-Methylbutyrylglycinuria | ACADSB |
| 3-Methylcrotonyl-CoA Carboxylase Deficiency | MCCC1, MCCC2 |
| 3-Methylglutaconic Aciduria Type I | AUH |
| 3-Phosphoglycerate Dehydrogenase Deficiency | PHGDH |
| Abetalipoproteinemia | MTTP |
| Argininemia | ARG1 |
| Argininocuccinic Aciduria | ASL |
| Biogenic Amines Synthesis Defect | GCH1 |
| Biotinidase Deficiency | BTD |
| Carnitine-Acylcarnitine Translocase Deficiency | SLC25A20 |
| Carnitine Palmitoyltransferase I Deficiency | CPT1A |
| Carnitine Palmitoyltransferase II Deficiency | CPT2 |
| Carnitine Uptake Deficiency/Carnitine Transport Defect | SLC22A5 |
| Creatine Deficiency Syndromes | GAMT, GATM |
| Cerebrotendinous Xanthomatosis | CYP27A1 |
| Citrullinemia Type I | ASS1 |
| Citrin Deficiency | SLC25A13 |
| Classic Galactosemia | GALT |
| Combined Pituitary Hormone Deficiency | LHX3, PROP1 |
| Congenital Disorders of Glycosylation Type Ib | MPI |
| Corticosterone Methyloxidase Deficiency | CYP11B2 |
| Crigler-Najjar Syndrome | UGT1A1 |
| Cystinosis | CTNS |
| Fabry Disease | GLA |
| Galactose Epimerase Deficiency | GALE |
| Galactokinase Deficiency | GALK1 |
| Glucose-6-Phosphate Dehydrogenase Deficiency | G6PD |
| Glutaric Acidemia Type I | GCDH |
| Glutaric Acidemia Type II | ETFA, ETFB, ETFDH |
| Glycogen Storage Disease IIa | G6PC |
| Glycogen Storage Disease IIIa | AGL |
| Glycogen Storage Disease 0 (GSD 0) | GYS2 |
| Glycogen Storage Disease IIb (GSD IIb) | SLC37A4 |
| Glycogen Storage Disease II (Pompe Disease) | GAA |
| Glycogen Storage Disease VI | PYGL |
| Hereditary Fructose Intolerance | ALDOB |
| HMG-CoA Lyase Deficiency | HMGCL |
| Holocarboxylase Synthetase Deficiency | HLCS |
| Homocystinuria | CBS |
| Homocystinuria (Cobalamin Disorders) | MMADHC, MTR, MTRR |
| Hypercholesterolemia | LDLR |
| Hypermethioninemia | AHCY, GNMT, MAT1A |
| Hypophosphatasia | ALPL |
| Isovaleric Acidemia | ACAD8 |
| Isovaleryl-CoA Dehydrogenase Deficiency | IVD |
| Krabbe Disease | GALC |
| Lipoprotein Lipase Deficiency (LPL) | LPL |
| Long-chain L-3-Hydroxyacyl-CoA Dehydrogenase Deficiency (LCHAD Deficiency) | HADHA |
| Lysinuric Protein Intolerance | SLC7A7 |
| Lysosomal Acid Lipase Deficiency | LIPA |
| Malonyl-CoA Decarboxylase Deficiency | MLYCD |
| Maple Syrup Urine Disease | BCKDHA, BCKDHB, DBT |
| Maple Syrup Urine Disease III | DLD |
| 3-Hydroxyacyl-CoA Dehydrogenase Deficiency | HADH |
| Medium-chain Acyl-CoA Dehydrogenase Deficiency | ACADM |
| Metachromatic Leukodystrophy | ARSA |
| Methylmalonic Acidemia (Cobalamin Disorders) | MMAA, MMAB, MMADHC |
| Methylmalonic Acidemia with Homocystinuria | ABCD4, LMBRD1, MMACHC, MMADHC, HCFC1 |
| Methylmalonic Acidemia, mut(0) Type | MMUT |
| Methylmalonyl-CoA Epimerase Deficiency | MCEE |
| Mucopolysaccharidosis Type I | IDUA |
| Mucopolysaccharidosis II (Hunter Syndrome) | IDS |
| N-Acetylglutamate Synthase Deficiency | NAGS |
| Nephrogenic Diabetes Insipidus Type II | AQP2 |
| Niemann-Pick Disease Type A/B | SMPD1 |
| Niemann-Pick Disease Type C1/D | NPC1 |
| Ornithine Transcarbamylase Deficiency | OTC |
| Ornithine Translocase Deficiency (Hyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome) | SLC25A15 |
| Phenylalanine Hydroxylase Deficiency (Phenylketonuria) | PAH |
| Primary Hyperoxaluria Type I | AGXT |
| Primary Hyperoxaluria Type II | GRHPR |
| Primary Hyperoxaluria Type III | HOGA1 |
| Propionic Acidemia | PCCA, PCCB |
| Short-chain Acyl-CoA Dehydrogenase Deficiency | ACADS |
| α-Methylacetoacetic Aciduria | ACAT1 |
| Transient Infantile Liver Failure | TRMU |
| Trifunctional Protein Deficiency | HADHA, HADHB |
| Segawa Syndrome | TH |
| Tyrosinemia Type I | FAH |
| Tyrosinemia Type II | TAT |
| Tyrosinemia Type III | HPD |
| Very Long-chain Acyl-CoA Dehydrogenase Deficiency (VLCAD) | ACADVL |
| Wilson Disease | ATP7B |
| X-linked Adrenoleukodystrophy | ABCD1 |
| Other: Genetic, Immunodeficiency, Pulmonary, Musculoskeletal | Gene |
|---|---|
| Cystic Fibrosis | CFTR |
| Severe Combined Immunodeficiency | ADA, IL7R, JAK3, IL2RG |
| Immunodeficiency 14A/B | PIK3CD |
Cardiomyopathy Kit
| Disorder | Gene |
|---|---|
| Arrhythmogenic Cardiomyopathy (ACM)/Arrhythmogenic Right Ventricular Cardiomyopathy (ARVC) | CTNNA3 |
| DES | |
| DSC2 | |
| DSG2 | |
| DSP | |
| FLNC | |
| JUP | |
| LMNA | |
| PKP2 | |
| PLN | |
| RYR2 | |
| TGFB3 | |
| TMEM43 | |
| TTN | Dilated Cardiomyopathy | ABCC9 |
| ACTC1 | |
| ACTN2 | |
| ANKRD1 | |
| BAG3 | |
| CASQ2 | |
| CRYAB | |
| CSRP3 | |
| DES | |
| DMD | |
| DSC2 | |
| DSG2 | |
| DSP | |
| DTNA | |
| EMD | |
| FKTN | |
| FLNC | |
| GATAD1 | |
| ILK | |
| JPH2 | |
| JUP | |
| LAMA4 | |
| LAMP2 | |
| LDB3 | |
| LMNA | |
| MYBPC3 | |
| MYH6 | |
| MYH7 | |
| MYL2 | |
| MYL3 | |
| MYPN | |
| NEBL | |
| NEXN | |
| PDLIM3 | |
| PKP2 | |
| PLN | |
| PRDM16 | |
| RAF1 | |
| RBM20 | |
| RYR2 | |
| SCN5A | |
| SGCD | |
| TAFAZZIN | |
| TCAP | |
| TMEM43 | |
| TNNC1 | |
| TNNI3 | |
| TNNT2 | |
| TPM1 | |
| TTN | |
| TTR | |
| VCL | |
| Hypertrophic Cardiomyopathy(HCM) | AARS2 |
| ABCC9 | |
| ACAD9 | |
| ACADVL | |
| ACTA1 | |
| ACTC1 | |
| AGK | |
| ALMS1 | |
| ALPK3 | |
| Cardiomyopathy occurring in Infancy or Childhood | BAG3 |
| COA5 | |
| COA6 | |
| COX15 | |
| CPT2 | |
| CRYAB | |
| DNAJC19 | |
| DOLK | |
| DSP | |
| ELAC2 | |
| FLNC | |
| GAA | |
| GBE1 | |
| GLA | |
| GTPBP3 | |
| HADHA | |
| HADHB | |
| HRAS | |
| KARS1 | |
| LAMP2 | |
| MRPL3 | |
| MRPL44 | |
| MTO1 | |
| MYBPC3 | |
| MYH7 | |
| MYPN | |
| NDUFB11 | |
| NDUFV2 | |
| PPA2 | |
| PRKAG2 | |
| RAF1 | |
| SCO2 | |
| SLC22A5 | |
| SLC25A20 | |
| SLC25A3 | |
| SLC25A4 | |
| TAFAZZIN | |
| TK2 | |
| TMEM70 | |
| TNNI3 | |
| TNNT2 | |
| TPM1 | |
| TSFM | |
| Left Ventricular Noncompaction Cardiomyopathy(NCCM/LVNC) | ACTC1 |
| DTNA | |
| HCN4 | |
| LDB3 | |
| LMNA | |
| MIB1 | |
| MYBPC3 | |
| MYH7 | |
| PKP2 | |
| PRDM16 | |
| TAFAZZIN | |
| TNNT2 | |
| TPM1 | |
| TTN | |
| Restrictive Cardiomyopathy | ACTC1 |
| BAG3 | |
| CRYAB | |
| DES | |
| FLNC | |
| MYBPC3 | |
| MYH7 | |
| MYL2 | |
| MYL3 | |
| MYPN | |
| TNNI3 | |
| TNNT2 | |
| TPM1 | |
| TTR | |
| Hypertrophic Cardiomyopathy(HCM) | ACTC1 |
| ACTN2 | |
| AKAP9 | |
| ANKRD1 | |
| CAV3 | |
| CRYAB | |
| CSRP3 | |
| DES | |
| FHL1 | |
| FLNC | |
| GAA | |
| GLA | |
| JPH2 | |
| LAMP2 | |
| LDB3 | |
| MYBPC3 | |
| MYH6 | |
| MYH7 | |
| MYL2 | |
| MYL3 | |
| MYLK2 | |
| MYOZ2 | |
| MYPN | |
| NEXN | |
| PLN | |
| PRKAG2 | |
| TCAP | |
| TNNC1 | |
| TNNI3 | |
| TNNT2 | |
| TPM1 | |
| TTN | |
| TTR | |
| VCL |
Arrhythmia Kit
| Disorder | Gene |
|---|---|
| Brugada Syndrome (BrS) | ABCC9 |
| CACNA1C | |
| CACNA2D1 | |
| CACNB2 | |
| GPD1L | |
| HCN4 | |
| KCNAB2 | |
| KCND3 | |
| KCNE3 | |
| KCNH2 | |
| KCNJ8 | |
| RYR2 | |
| SCN10A | |
| SCN1B | |
| SCN2B | |
| SCN3B | |
| SCN5A | |
| TRPM4 | |
| Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT) | ANK2 |
| CALM1 | |
| CALM2 | |
| CALM3 | |
| CASQ2 | |
| GNAI2 | |
| HCN4 | |
| KCNJ2 | |
| KCNQ1 | |
| LMNA | |
| RYR2 | |
| SCN5A | |
| TECRL | |
| TRDN | |
| Long QT Syndrome (LQTS) | AKAP9 |
| ANK2 | |
| CACNA1C | |
| CALM1 | |
| CALM2 | |
| CALM3 | |
| CAV3 | |
| KCNE1 | |
| KCNE2 | |
| KCNH2 | |
| KCNJ2 | |
| KCNJ5 | |
| KCNQ1 | |
| RYR2 | |
| SCN4B | |
| SCN5A | |
| SNTA1 | |
| TECRL | |
| TRDN | |
| Atrial Fibrillation and Short QT Syndrome | ABCC9 |
| CACNA1C | |
| CACNA2D1 | |
| CACNB2 | |
| GJA5 | |
| HCN4 | |
| KCNA5 | |
| KCNE1 | |
| KCNE2 | |
| KCNH2 | |
| KCNJ2 | |
| KCNQ1 | |
| LMNA | |
| MYL4 | |
| NPPA | |
| SCN1B | |
| SCN2B | |
| SCN3B | |
| SCN4B | |
| SCN5A | |
| TBX5 | |
| TNNI3 |
Aortic Disorders Kit
| Disorders | Genes |
|---|---|
| Aortic Disorders (Including Marfan Syndrome, Ehlers-Danlos Syndrome, Loeys-Dietz Syndrome, Familial Thoracic Aortic Aneurysm and Dissection, Mitral Valve Diseases, etc.) |
ABCC6 |
| ACTA2 | |
| ACVR1 | |
| ADAMTS2 | |
| ALDH18A1 | |
| ATP6V0A2 | |
| ATP6V1A | |
| ATP6V1E1 | |
| B3GALT6 | |
| B4GALT7 | |
| BGN | |
| C1R | |
| C1S | |
| CBS | |
| CHST14 | |
| COL1A1 | |
| COL1A2 | |
| COL3A1 | |
| COL4A1 | |
| COL5A1 | |
| COL5A2 | |
| DSE | |
| EFEMP2 | |
| ELN | |
| FBLN5 | |
| FBN1 | |
| FBN2 | |
| FKBP14 | |
| FOXE3 | |
| GORAB | |
| LOX | |
| LTBP4 | |
| MFAP5 | |
| MYH11 | |
| MYLK | |
| PLOD1 | |
| PRDM5 | |
| PYCR1 | |
| SKI | |
| SLC2A10 | |
| SLC39A13 | |
| SMAD3 | |
| SMAD4 | |
| TGFB2 | |
| TGFB3 | |
| TGFBR1 | |
| TGFBR2 | |
| ZNF469 |
Congenital Heart Defects Kit
| Disorders | Genes |
|---|---|
| Isolated and Syndromic Congenital Heart Diseases (Including Heterotaxy, Alagille Syndrome, Ventricular Septal Defect, Aortic Valve Stenosis, Tetralogy of Fallot, etc.) |
ACTA2 |
| ACTC1 | |
| ACVR2B | |
| AFF4 | |
| BMPR2 | |
| CAD | |
| CFAP53 | |
| CDK13 | |
| CELSR1 | |
| CELSR2 | |
| CELSR3 | |
| CHD4 | |
| CHD7 | |
| CITED2 | |
| CREBBP | |
| CRELD1 | |
| DNAH11 | |
| DNAH5 | |
| DNAH6 | |
| DNAI1 | |
| DTNA | |
| EHMT1 | |
| ELN | |
| EVC | |
| EVC2 | |
| FBN1 | |
| FLNA | |
| FOXC1 | |
| FOXH1 | |
| GANAB | |
| GATA4 | |
| GATA5 | |
| GATA6 | |
| GDF1 | |
| GPC3 | |
| HAND1 | |
| HAND2 | |
| HRAS | |
| JAG1 | |
| KDM5B | |
| KMT2D | |
| MED13L | |
| MMP21 | |
| MYH11 | |
| MYH6 | |
| MYH7 | |
| NIPBL | |
| NKX2-5 | |
| NKX2-6 | |
| NME7 | |
| NODAL | |
| NOTCH1 | |
| NOTCH2 | |
| NR2F2 | |
| PITX2 | |
| PKD1L1 | |
| PLD1 | |
| POGZ | |
| PRDM6 | |
| PRKD1 | |
| RABGAP1L | |
| RBFOX2 | |
| RBM10 | |
| SALL4 | |
| SEMA3D | |
| SEMA3E | |
| SMAD6 | |
| TAB2 | |
| TBX1 | |
| TBX20 | |
| TBX5 | |
| TFAP2B | |
| TGFBR1 | |
| TGFBR2 | |
| TLL1 | |
| TMEM260 | |
| TPM1 | |
| ZEB2 | |
| ZFPM2 | |
| ZIC3 |
FH, PH, RASopathies Kit
| Disorder | Genes |
|---|---|
| RASopathies (Cardiofaciocutaneous syndrome, Costello syndrome, Legius syndrome, Neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, Noonan-like syndrome with loose anagen hair, etc.) |
AKT3 |
| BRAF | |
| CBL | |
| CCND2 | |
| EPHB4 | |
| HRAS | |
| KRAS | |
| LZTR1 | |
| MAP2K1 | |
| MAP2K2 | |
| MRAS | |
| NF1 | |
| NF2 | |
| NRAS | |
| PIK3CA | |
| PIK3R2 | |
| PPP1CB | |
| PTPN11 | |
| RAF1 | |
| RASA1 | |
| RASA2 | |
| RIT1 | |
| RRAS | |
| SASH1 | |
| SHOC2 | |
| SMARCB1 | |
| SOS1 | |
| SOS2 | |
| SPRED1 | |
| STAMBP | |
| Familial Hypercholesterolemia (FH) | ABCA1 |
| ABCG5 | |
| ABCG8 | |
| APOA5 | |
| APOB | |
| APOE | |
| LDLR | |
| LDLRAP1 | |
| LIPA | |
| LPL | |
| PCSK9 | |
| Pulmonary Hypertension (PH) | ACVRL1 |
| BMPR1B | |
| BMPR2 | |
| CAV1 | |
| EIF2AK4 | |
| ENG | |
| KCNA5 | |
| KCNK3 | |
| SMAD4 | |
| SMAD9 | |
| TBX4 |
Metabolic Kit
*The list of disorders above is not comprehensive
| Disorder | Gene |
|---|---|
| Hyperinsulinemic Hypoglycemia | ABCC8 |
| Adrenoleukodystrophy | ABCD1 |
| Methylmalonic Acidemia and Homocystinuria, cblJ Type | ABCD4 |
| Isobutyryl-CoA Dehydrogenase Deficiency (also known as IBD Deficiency, ACAD8 Deficiency) | ACAD8 |
| Complex I Deficiency, Nuclear Type 20 (also known as Mitochondrial Complex I Deficiency due to ACAD9 Deficiency, ACAD9 Deficiency) | ACAD9 |
| Medium Chain Acyl-CoA Dehydrogenase (MCAD) Deficiency (also known as ACADM Deficiency, MCAD Deficiency, MCADH Deficiency, Medium Chain Acyl-CoA Dehydrogenase Deficiency) | ACADM |
| Short Chain Acyl-CoA Dehydrogenase (SCHAD) Deficiency (also known as ACADS Deficiency, Short Chain Acyl-CoA Dehydrogenase Deficiency, SCADH Deficiency, SCAD Deficiency) | ACADS |
| Short/Branched Chain Acyl-CoA Dehydrogenase Deficiency - SBCADD (also known as 2-Methylbutyryl-CoA Dehydrogenase Deficiency, 2-Methylbutyrylglycinuria) | ACADSB |
| Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficiency (also known as VLCAD Deficiency) | ACADVL |
| Peroxisomal Acyl-CoA Oxidase Deficiency (also known as Straight-Chain Acyl-CoA Oxidase Deficiency, Pseudo-Adrenoleukodystrophy) | ACOX1 |
| Mixed Malonic and Methylmalonic Acidemia | ACSF3 |
| Aspartylglucosaminuria | AGA |
| Glycogen Storage Disease III (a and b) (also known as Forbes Disease, Cori Disease, Limit Dextrinosis, Amylo-1,6-Glucosidase Deficiency, AGL Deficiency) | AGL |
| Chondrodysplasia Punctata, Type 3 (also known as Alkylglycerone Phosphate Synthase Deficiency, Alkylglycerone Phosphate Synthetase Deficiency, AGPS Deficiency) | AGPS |
| Methylmalonate Semialdehyde Dehydrogenase Deficiency (also known as MMSDH Deficiency) | ALDH6A1 |
| Glycogen Storage Disease XII (also known as GSD XII, Aldolase A Deficiency, ALDOA Deficiency, Aldolase Deficiency - Erythrocyte, Erythrocyte Aldolase Deficiency) | ALDOA |
| Fructose Intolerance, Hereditary (also known as Fructosemia, Fructose-1-Phosphate Aldolase Deficiency, Fructose-1,6-Bisphosphate Aldolase B Deficiency, Aldolase B Deficiency, ALDOB Deficiency) | ALDOB |
| Constitutional Disorders of Glycosylation, Ik Type | ALG1 |
| Constitutional Disorders of Glycosylation, Ip Type | ALG11 |
| Constitutional Disorders of Glycosylation, Ig Type | ALG12 |
| Constitutional Disorders of Glycosylation, I Type (also known as Developmental and Epileptic Encephalopathy 36, Epileptic Encephalopathy, Infantile, 36) | ALG13 |
| Constitutional Disorders of Glycosylation, II Type | ALG2 |
| Constitutional Disorders of Glycosylation, Id Type (also known as CDG Id - CDGId, Congenital Disorder of Glycosylation IV, formerly CDGS4) | ALG3 |
| Constitutional Disorders of Glycosylation Ic Type (also known as CDG-Ic with defective glycosylation of dolichol linked oligosaccharides, Congenital Disorder of Glycosylation Type I, formerly Glycosylation Defects Type V, CDGS5 Type) | ALG6 |
| Constitutional Disorders of Glycosylation Ih - CDG1H Type | ALG8 |
| Constitutional Disorders of Glycosylation I1 - CDGI1 Type | ALG9 |
| Gillessen-Kaesbach-Nishimura Syndrome (also known as Multiple Cystic Kidneys, Microcephaly and Short Stature Autosomal Recessive, Microcephaly and Short Stature) | |
| α-Methylacyl-CoA Racemase Deficiency (AMACR Deficiency) | AMACR |
| Constitutional Bile Acid Synthesis Defect 4 (also known as Intrahepatic Cholestasis with Tricarboxylic Aciduria, Tricarboxylic Acid in Bile) | |
| Glycine Encephalopathy (also known as Non-Ketotic Hyperglycinemia - NKH) | AMT |
| Argininemia (also known as Arginase Deficiency, Hyperargininemia, ARG1 Deficiency) | ARG1 |
| Metachromatic Leukodystrophy - MLD (also known as Metachromatic Leukodystrophy, Sulfatide Lipidosis, Arylsulfatase A Deficiency, ARSA Deficiency, Cerebroside Sulfatase Deficiency) | ARSA |
| Mucopolysaccharidosis VI Type (Maroteaux-Lamy) (also known as MPS VI, Maroteaux-Lamy Syndrome, Arylsulfatase B Deficiency, ARSB Deficiency, N-Acetylgalactosamine-4-Sulfatase Deficiency) | ARSB |
| Argininocuccinate Lyase Deficiency (also known as Argininosuccinate Lyase Deficiency, ASL Deficiency, Argininosuccinase Deficiency, ASP Deficiency, Aminoacylase 2 Deficiency, ACY2 Deficiency) | ASL |
| Canavan Disease (also known as Canavan-Van Bogaert-Bertrand Disease, Central Nervous System Spongiform Degeneration, Aspartoacylase Deficiency, ASPA Deficiency, ASP Deficiency, Aminoacylase 2 Deficiency, ACY2 Deficiency) | ASPA |
| Citrullinemia (also known as Citrullinemia I - CTLN1 Type, Citrullinuria, Argininosuccinate Synthetase Deficiency, ASS Deficiency) | ASS1 |
| 3-Methylglutaconic Aciduria, I-MGCA1 Type (also known as MGA Type I - MGA1, 3-Methylglutaconyl-CoA Hydratase Deficiency, 3-MG-CoA Hydratase Deficiency) | AUH |
| Constitutional Disorders of Glycosylation IId Type | B4GALT1 |
| Maple Syrup Urine Disease IIa Type (also known as Branched-Chain Ketoaciduria, Branched-Chain α-Keto Acid Dehydrogenase Deficiency, BCDK Deficiency, Keto Acid Decarboxylase Deficiency) | BCKDHA |
| Maple Syrup Urine Disease Ib Type (also known as Branched-Chain Ketoaciduria, Branched-Chain α-Keto Acid Dehydrogenase Deficiency, BCKD Deficiency, Keto Acid Decarboxylase Deficiency) | BCKDHB |
| Biotinidase Deficiency (also known as BTD Deficiency, Late-Onset Multiple Carboxylase Deficiency, Early-Onset Multiple Carboxylase Deficiency) | BTD |
| Developmental and Epileptic Encephalopathy 50 (also known as Epileptic Encephalopathy, Early Infantile, 50 - EIEE50, Glyxosylation type Iz Congenital Disorder, former CDG1Z) | CAD |
| Constitutional Disorders of Glycosylation IIo Type | CCDC115 |
| Transient Methylmalonic Aciduria due to Transcobalamin Receptor Defect (also known as Methylmalonic Acidemia, TCblR Type) | CD320 |
| Neuronal Ceroid Lipofuscinosis 3 (also known as Juvenile Neuronal Ceroid Lipofuscinosis, Batten Disease, Spielmeyer-Sjögren Disease) | CLN3 |
| Neuronal Ceroid Lipofuscinosis 5 (also known as Neuronal Ceroid Lipofuscinosis 5, Onset Age Variability) | CLN5 |
| Neuronal Ceroid Lipofuscinosis 6A (also known as Neuronal Ceroid Lipofuscinosis, Late Infantile Variant vLINCL) | CLN6 |
| Neuronal Ceroid Lipofuscinosis 6B | |
| Neuronal Ceroid Lipofuscinosis 8 | CLN8 |
| Neuronal Ceroid Lipofuscinosis 8 Type, Northern Epilepsy Variant (also known as Progressive Epilepsy with Mental Retardation) | |
| 3-Methylglutaconic Aciduria VIIA Type (also known as Neurologic Disorder and Neutropenia with 3-Methylglutaconic Aciduria) | CLPB |
| 3-Methylglutaconic Aciduria VIIB Type (also known as MGCA7, Cataract with 3-Methylglutaconic Aciduria, Neurologic Disorder and Neutropenia - MEGCANN) | |
| Severe Congenital Neutropenia 9 | |
| Constitutional Disorders of Glycosylation IIg Type | COG1 |
| Constitutional Disorders of Glycosylation Iq - CDG Iq Type | COG2 |
| Constitutional Disorders of Glycosylation IIijcdg IIj Type | COG4 |
| Wilson's Disease (also known as Cerebellar Ataxia) | |
| Constitutional Disorders of Glycosylation IIIiCDG IIi Type | COG5 |
| Constitutional Disorders of Glycosylation IIIlCDG III Type | COG6 |
| Shohat Syndrome | |
| Constitutional Disorders of Glycosylation IIe Type | COG7 |
| Constitutional Disorders of Glycosylation IIh-CDG IIh Type | COG8 |
| Carbamoyl Phosphate Synthetase I Deficiency (also known as CPS I Deficiency) | CPS1 |
| Carnitine Palmitoyltransferase I Deficiency (also known as CPT I Deficiency, CPT Deficiency, Hepatic, Type I) | CPT1A |
| Carnitine Palmitoyltransferase II Deficiency, Infantile (also known as Carnitine Palmitoyltransferase II Deficiency with Ketotic Hypoglycemia, Carnitine Palmitoyltransferase II Deficiency, Hepatic, Muscle, CPT II Deficiency, Liver, CPT2 Deficiency, Infantile) | CPT2 |
| Carnitine Palmitoyltransferase II Deficiency, Lethal Neonatal (also known as Carnitine Palmitoyltransferase II Deficiency Neonatal, CPT II Deficiency Neonatal, CPT2 Deficiency Neonatal) | |
| Carnitine Palmitoyltransferase II Deficiency, Myopathic, Stress-Induced (also known as Carnitine Palmitoyltransferase II Deficiency, Adult Onset, CPT II Deficiency, CPT2 Deficiency, Late-Onset) | |
| Cystinosis, Non-Nephropathic or Nephropathic Variant (also known as Lysosomal Cystine Transporter Deficiency, Cystinosin Deficiency) | CTNS |
| Cystinosis, Late-Onset Juvenile or Adolescent Nephropathic (also known as Intermediate Cystinosis) | |
| Cystinosis, Adult Non-Nephropathic (also known as Non-Nephropathic Cystinosis with Ocular Manifestations, Benign Non-Nephropathic Cystinosis) | |
| Galactosialidosis (also known as Goldberg Syndrome, Neuraminidase Deficiency with Beta-Galactosidase Deficiency, Neuraminidase/Beta-Galactosidase Expression - NGBE, Protective Protein-Cathepsin A Deficiency, PPCA Deficiency) | CTSA |
| Gonadal Dysgenesis (also known as Gonadal Dysgenesis, PYCD) | CTSK |
| Maple Syrup Urine Disease, Type II (also known as Branched-Chain Ketoaciduria, Branched-Chain α-Keto Acid Dehydrogenase Deficiency, BCKD Deficiency, Keto Acid Decarboxylase Deficiency) | DBT |
| Constitutional Disorders of Glycosylation, IrCDG Ir Type | DDOST |
| Smith-Lemli-Opitz Syndrome (also known as SLO Syndrome, RSH Syndrome, Rytledge Lethal Multiple Congenital Anomaly Syndrome, Polydactyly, Sexual Reversal Renal Anomalies, Single Lung Congenital Syndrome) | DHCR7 |
| Constitutional Disorders of Glycosylation 1bb-Retinitis Pigmentosa 59 Type | DHDDS |
| Developmental Delay and Seizures, with or without Movement Abnormalities | |
| Maple Syrup Urine Disease, Type III (also known as Dihydrolipoyl Amide Dehydrogenase Deficiency, DLD Deficiency) | DLD |
| Mild Hyperphenylalaninemia, Non-BH4-Deficient | DNAJC12 |
| 3-Methylglutaconic Aciduria, Type V-MGCA5 (also known as Dilated Cardiomyopathy with Ataxia (DCMA)) | DNAJC19 |
| Constitutional Disorders of Glycosylation Im Type (also known as Dolichol Kinase Deficiency, DK1 Deficiency) | DOLK |
| Constitutional Disorders of Glycosylation Ij-CDG Ij Type | DPAGT1 |
| Thirteen Cases of Congenital Myasthenic Syndrome with Tubular Aggregates (also known as Congenital Myasthenic Syndrome 2- CMSTA2) | DPAGT1 |
| Constitutional Disorders of Glycosylation, IeCDG Ie Type | DPM1 |
| Constitutional Disorders of Glycosylation IuCDG Iu Type | DPM2 |
| Muscular Dystrophy-Dystroglycanopathy (Congenital with Intellectual Disability), B Type, 15 Type (also known as Congenital DPM3-Related Muscular Dystrophy) | DPM3 |
| Muscular Dystrophy-Dystroglycanopathy (Limb-Girdle), C Type, 15-MDDGC15 (also known as Muscular Dystrophy-Dystroglycanopathy, Limb-Girdle DPM3-Related Congenital Glycosylation Type Io-CDG1O, CDG Io, CDG1) | |
| Glycogen Storage Disease XIII (13) (also known as GSD XIII, Enolase 3 Deficiency, Enolase Beta Deficiency) | ENO3 |
| Multiple Acyl-CoA Dehydrogenase Deficiency, MADD (also known as Glutaric Acidemia IIA, Glutaric Aciduria IIA, GA2A, Ethylmalonic-Adipic Aciduria - EMA) | ETFA |
| Multiple Acyl-CoA Dehydrogenase Deficiency, MADD (also known as Glutaric Acidemia IIB, Glutaric Aciduria IIB, GA2B, Ethylmalonic-Adipic Aciduria - EMA) | ETFB |
| Multiple Acyl-CoA Dehydrogenase Deficiency, MADD (also known as Glutaric Acidemia IIC, Glutaric Aciduria IIC, GA2C, Ethylmalonic-Adipic Aciduria-EMA) | ETFDH |
| Fructose-1,6-Bisphosphatase Deficiency | FBP1 |
| Fucosidosis (also known as α-L-Fucosidase Deficiency) | FUCA1 |
| Glycosylation Disorders with Fucosylation 1-CDGFF | FUT8 |
| Glycogen Storage Disease IIa (GSD II, Acid Maltase Deficiency, GAA Deficiency, Pompe Disease, Generalized Glycogenosis, Cardiomegalic Form, Diffuse Glycogenosis, Acid Maltase Deficiency-AMD, α-1,4-Glucosidase Deficiency) | GAA |
| Krabbe Disease (also known as Globoid Cell Leukodystrophy-GLD, Globoid Cell Leukodystrophy, Galactosylceramidase Beta-Galactosidase Deficiency, Galactosylceramide Beta-Galactosidase Deficiency, Galc Deficiency) | GALC |
| Mucopolysaccharidosis IVA (also known as Morquio Syndrome A, MPS IVA, Morquio A Disease, Galactosamine-6-Sulfatase Deficiency, GALNS Deficiency) | GALNS |
| Creatine Deficiency Syndromes 2-CCDS2 (also known as Guanidinoacetate Methyltransferase Deficiency, GAMT Deficiency, Creatine Deficiency due to AGAT Deficiency) | GAMT |
| Creatine Deficiency Syndromes 3-CCDS3 (also known as Arginine:Glycine Amidinotransferase Deficiency, AGAT Deficiency, GATM Deficiency, Creatine Deficiency due to AGAT Deficiency) | GATM |
| Gaucher Disease, Perinatal Lethal (also known as Gaucher Disease, Congenital Type) | GBA |
| Gaucher Disease, Type I (also known as GD I, Gaucher Disease, Non-Neuronopathic Juvenile, Glucosylceramidase Deficiency, Acid β-Glucosidase Deficiency, GBA Deficiency) | |
| Gaucher Disease, Type II (also known as Gaucher Disease, Acute Neurologic Type) | |
| Gaucher Disease, Type III (also known as Gaucher Disease, Subacute Neuronopathic Type, Gaucher Disease, Chronic Neurologic Type, Gaucher Disease, Juvenile and Adult Brain) | |
| Gaucher Disease | |
| Glycogen Storage Disease IV (also known as GSD IV, Glycogen Branching Enzyme Deficiency, GBE1 Deficiency, Andersen Disease, Brancher Deficiency, Glycogenosis IV, Amylopectinosis, Cirrhosis, Familial, Abnormal Glycogen Body Deposit Family) | GBE1 |
| Polysaccharide Storage Diseases, Adult Onset (also known as Adult Polyglucosan Body Disease-APBN) | |
| Glutaric Acidemia I (also known as Glutaric Acidemia I-GA1, Glutaric Aciduria I, GA I, Glutaryl-CoA Dehydrogenase Deficiency) | GCDH |
| Hyperphenylalaninemia, BH4 Deficiency, B-HPAH4B (also known as GTP Cyclohydrolase I Deficiency, GTPCH1 Deficiency) | GCH1 |
| Dystonia, Dopa-Responsive, with or without Hyperphenylalaninemia (also known as Dystonia Dopa-Responsive-DRD, Dystonia 5-DYT5, Dystonia Progressive with Diurnal Variation, Dystonia-Parkinsonism with Diurnal Variation, Segawa Syndrome Autosomal Dominant, Dystonia Dopa-Responsive Autosomal Dominant, Dystonia Dopa-Responsive Autosomal Dominant with Diurnal Variation) | |
| Hyperinsulinism, Familial, 3 | GCK |
| Glycine Encephalopathy-GCE (also known as Non-Ketotic Hyperglycinemia-NKH) | GCSH |
| Fabry Disease (also known as Diffuse Angiokeratoma, Anderson-Fabry Disease, Hereditary Dystopic Lipidosis, α-Galactosidase A Deficiency, GLA Deficiency, Ceramide Trihexosidase Deficiency) | GLA |
| GM1 Gangliosidosis, Type I (also known as Generalized GM1 Gangliosidosis Type I, Systemic GM1 Gangliosidosis, β-Galactosidase-1 Deficiency, GLB1 Deficiency) | GLB1 |
| GM1 Gangliosidosis, Type II (also known as Systemic GM1 Juvenile Type, Systemic GM1 Type) | |
| GM1 Gangliosidosis, Type III (also known as Generalized GM1 Gangliosidosis Adult Type, Generalized GM1 Gangliosidosis Chronic Type, Generalized GM1 Type 3) | GLB1 |
| Mucopolysaccharidosis IVB (Morquio Syndrome B, MPS IVB) | |
| Glycine Encephalopathy (also known as Non-Ketotic Hyperglycinemia-NKH) | GLDC |
| Hyperinsulinism and Hyperammonemia Syndrome (also known as Familial Hyperinsulinemic Hypoglycemia 6-HHF6) | GLUD1 |
| GM2 Gangliosidosis, AB Variant (also known as Hexosaminidase A Activator Deficiency, GM2 Activator Deficiency, AB Variant GM2 Gangliosidosis, Tay-Sachs Disease AB Variant) | GM2A |
| Araldimaschia, Achalasia, Mental Retardation Syndrome | GMPPA |
| Nonaka Myopathy (also known as Nonaka Distal Myopathy, Distal Myopathy with or without Rimmed Vacuoles, Inclusion Body Hereditary Myopathy, Autosomal Recessive IBM, Rimmed Vacuolar Myopathy, Quadriceps Sparing Myopathy, GNE Myopathy, Inclusion Body Myopathy, Formerly IBM2 Autosomal Recessive) | GNE |
| Sialuria (also known as Sialuria French Type) | |
| Mucolipidosis IIIC Alpha/Beta (also known as I Cell Disease-ICD) | GNPTAB |
| Mucolipidosis IIID Alpha/Beta (also known as Pseudo-Hurler Polydystrophy) | |
| Mucolipidosis IIIGamma | GNPTG |
| Mucopolysaccharidosis IIID Type (also known as N-Acetylglucosamine-6-Sulfatase Deficiency, Sanfilippo Syndrome D) | GNS |
| Mucopolysaccharidosis VII (MPS VII, Sly Syndrome, β-Glucuronidase Deficiency, GUSB Deficiency) | GUSB |
| Glycogen Storage Disease XV (also known as GSD XV, Glycogenosis Type, GYG1 Deficiency) | GYG1 |
| Polysaccharide Myopathy 2 | |
| Glycogen Storage Disease 0, Muscle (also known as GSD 0b, Muscle Glycogen Storage Disease 0, Muscle Glycogen Synthase Deficiency) | GYS1 |
| Glycogen Storage Disease 0, Liver (also known as GSD 0a, Hepatic Glycogen Synthase Deficiency with Hypoglycemia, Hepatic Glycogen Synthase Deficiency, Hepatic Glycogen Storage Disease 0) | GYS2 |
| Familial Hyperinsulinism, 3 | HADH |
| 3-Hydroxyacyl-CoA Dehydrogenase Deficiency (also known as HADH Deficiency, SCHAD Deficiency) | |
| Long Chain 3-Hydroxyacyl-CoA Dehydrogenase (HADHA Deficiency) | HADHA |
| Mitochondrial Tri-Function Protein Deficiency (also known as Tri-Function Protein Deficiency) | |
| Mitochondrial Tri-Function Protein (TFP) Deficiency (also known as Tri-Function Protein Deficiency) | HADHB |
| Methylmalonic Acidemia and Homocystinuria, cblX Type (also known as Intellectual Disability, X-Linked 3, Mental Retardation X-Linked 3) | HCFC1 |
| Tay-Sachs Disease (also known as GM2 Gangliosidosis Type I, B Variant GM2 Gangliosidosis, Hexosaminidase A Deficiency, Hex A Deficiency) | HEXA |
| Sandhoff Disease (also known as GM2 Gangliosidosis Type II, Hexosaminidase A and B Deficiency) | HEXB |
| Mucopolysaccharidosis IIIC Type (Sanfilippo C) (also known as MPS IIIC, Sanfilippo Syndrome C, Acetyl-CoA:α-Glucosaminide N-Acetyltransferase Deficiency) | HGSNAT |
| Retinal Dystrophy 73 | |
| HMG-CoA Lyase Deficiency (also known as HMGCL Deficiency, HL Deficiency, Hydroxymethylglutaric Aciduria) | HMGCL |
| 3-Hydroxy-3-Methylglutaryl-CoA Synthase 2 (HMGCS2 Deficiency) | HMGCS2 |
| Tyrosinemia, Type III (also known as 4-Hydroxyphenylpyruvate Dioxygenase Deficiency, 4-Hydroxyphenylpyruvic Acid Oxidase Deficiency) | HPD |
| Hawkinsinuria | |
| Costello Syndrome (also known as Faciocutaneoskeletal Syndrome, FCS Syndrome, Congenital Myopathy with Excess Muscle Spindles) | HRAS |
| 2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency (also known as HSD17B10 Deficiency, 17-β-Hydroxysteroid Dehydrogenase X Deficiency, 3-Hydroxyacyl-CoA Dehydrogenase II Deficiency, 2-Methyl-3-Hydroxybutyryl-CoA Dehydrogenase Deficiency, MHBD Deficiency, Mental Retardation X-Linked Syndrome 10-MRXS10, Cerebral Ataxia and Mental Retardation with Abnormal Behavior CAMR, Cerebral Ataxia and Abnormal Behavior with Mental Retardation) | HSD17B10 |
| D-Bifunctional Protein Deficiency (also known as 17-β-Hydroxysteroid Dehydrogenase IV Deficiency, DBP Deficiency, Peroxisomal Bifunctional Enzyme Deficiency, PBFE Deficiency) | HSD17B4 |
| Mucopolysaccharidosis IX Type (also known as MPS IX, Hyaluronidase Deficiency) | HYAL1 |
| Mucopolysaccharidosis II Type (also known as MPS II, Hunter Syndrome, Iduronate 2-Sulfatase Deficiency, IDS Deficiency, Sulfoiduronate Sulfatase Deficiency, SIDS Deficiency) | IDS |
| Mucopolysaccharidosis Ih Type (also known as Hurler Syndrome) | IDUA |
| Mucopolysaccharidosis Ih/s Type (also known as Hurler-Scheie Syndrome) | |
| Mucopolysaccharidosis Is Type (also known as Scheie Syndrome, Formerly Mucopolysaccharidosis V Type, Formerly MPS V) | |
| Hyperinsulinemic Hypoglycemia, familial, 5 | INSR |
| Hyperinsulinemic Hypoglycemia, familial, 2-HF2 (other names: Infantile hyperinsulinism-PHHI persistent hyperinsulinemic hypoglycemia, persistent hypoglycemia, insulin-like syndrome by nest tumor, neonatal hyperinsulinism, congenital hyperinsulinism, familial hyperinsulinemia, neonatal hyperinsulinism) | KCNJ11 |
| Danon Disease (other: Former glycogen storage disease IIb) | LAMP2 |
| Glycogen Disease XI (other: Lactate dehydrogenase A deficiency, LDH subunit M) | LDHA |
| Hyperglycinemia, lactic acidosis, seizures-HGCLAS (Pyruvate dehydrogenase lipoyl synthetase deficiency-PDHLD) | LIAS |
| Lysosomal Acid Lipase Deficiency (other names: Wolman disease, cholesterol ester storage disease-CESD, LAL deficiency, cholesterol ester hydrolase deficiency) | LIPA |
| Methylmalonic Acidemia and Homocystinuria, cblF Type. (other names: Methylmalonic aciduria due to vitamin B12 release disorder) Vitamin B12 lysosomal release disorder. Cobalamin deficiency in lysosomal release of vitamin B12 storage disease Cobalamin F disease | LMBRD1 |
| Rafiq Syndrome | MAN1B1 |
| α-Mannosidosis (other names: Mannosidosis αB type lysosome MANSA, lysosomal α-D-mannosidase deficiency, α-mannosidase B deficiency) | MAN2B1 |
| β-Mannosidosis (other names: Mannosidase βA lysosome MANBA, MANB1, Mannanase, Mannanase) | MANBA |
| Methylmalonyl-CoA Epimerase Deficiency (other names: Methylmalonol-CoA racemase deficiency, formerly methylmalonic aciduria III) | MCEE |
| Mucolipidosis Type IV (other names: ML IV, Sialolipidosis) | MCOLN1 |
| Neuronal Ceroid Lipofuscinosis 7 | MFSD8 |
| Macular Dystrophy with Central Cone Infiltration | |
| Constitutional Glycosylation Disorder Type IIa (other names: CDG IIa, Alekraja syndrome, prominent coloboma and fissure, formerly carbohydrate-deficient glycoprotein syndrome type II, CDGS2) | MGAT2 |
| Malonyl-CoA Decarboxylase Deficiency | MLYCD |
| Methylmalonic Acidemia, cblA Type (other names: Methylmalonic aciduria due to adenosylcobalamin cblA synthesis defect vitamin B12 responsive) | MMAA |
| Methylmalonic Acidemia, cblB Type (other names: Methylmalonic aciduria due to adenosylcobalamin cblB synthesis defect vitamin B12 responsive) | MMAB |
| Methylmalonic Acidemia and Homocystinuria, cblC Type (other names: Methylmalonic aciduria and homocystinuria, vitamin B12 responsive). Vitamin B12 metabolic disorder with combined deficiency of methylmalonyl-CoA mutase and homocysteine: methyltetrahydrofolate methyltransferase | MMACHC |
| Methylmalonic Acidemia and Homocystinuria, cbID (other names: Methylmalonic aciduria and homocystinuria cblD type). Methylmalonic aciduria, cblH type Methylmalonic acidemia, cblH type previously | MMADHC |
| Constitutional Glycosylation Disorder, IIb Type (CDG IIb, glucosidase I deficiency) | MOGS |
| Constitutional Glycosylation Disorder, If Type (CDG If) | MPDU1 |
| Constitutional Glycosylation Disorder Ib Type (other names: CDG Ib, CDG Ib, CDG Gastrointestinal type, Mannose phosphate isomerase deficiency, MPI deficiency, Protein-losing enteropathy-hepatic fibrosis syndrome, Saguenay-Lac Saint-Jean syndrome, SLSJ syndrome) | MPI |
| Homocystinuria-Megaloblastic Anemia, cblG Type. (other names: Methylcobalamin deficiency cblG type) Methionine synthase deficiency | MTR |
| Homocystinuria-Megaloblastic Anemia, cblE Type. (other names: Vitamin B12-responsive homocystinuria cblE type) Methylcobalamin deficiency cblE type | MTRR |
| Methylmalonic Acidemia due to Methylmalonyl-CoA Mutase Deficiency (other name: Methylmalonic aciduria due to methylmalonyl-CoA mutase deficiency, MMA due to MCM deficiency, Methylmalonic aciduria mut type) | MMUT |
| Schindler Disease (other names: α-N-acetylgalactosaminidase (α-NAGA) deficiency) | NAGA |
| Mucopolysaccharidosis IIIB (Sanfilippo B) (other names: Mucopolysaccharidosis IIIB-MPS3B type, MPS IIIB type, Sanfilippo syndrome B type, n-acetyl-α-D-glucosaminidase deficiency, NAGLU deficiency) | NAGLU |
| N-Acetylglutamate Synthase (NAGS) Deficiency (other names: Hyperammonemia due to N-acetylglutamate synthetase deficiency, N-acetylglutamate synthetase deficiency, NAGS deficiency) | NAGS |
| Sialidosis Type I and II (other names: Sialidosis type I, Mucolipidosis type I, ML I, Lipomucopolysaccharidosis, Sialidase deficiency, Glycoprotein neuraminidase deficiency, NEUG deficiency, Neuraminidase 1 deficiency, NEU deficiency) | NEU1 |
| Congenital Disorder of Deglycosylation 1-CDDG1 (other names: Congenital disorder of deglycosylation-CDDG, previously Congenital disorder of glycosylation type IIv-CDG1V) | NGLY1 |
| Niemann-Pick Disease, C1/D Type (other names: Niemann-Pick disease C-NPC type, Niemann-Pick disease with cholesterol esterification block, Niemann-Pick disease subacute juvenile, Niemann-Pick disease chronic neurological type, Niemann-Pick disease without sphingomyelinase deficiency, Neurovisceral storage disorder with vertical gaze palsy) | NPC1 |
| Niemann-Pick Disease, C2 Type | NPC2 |
| Congenital Disorder of Glycosylation, Type 1aa | NUS1 |
| Seizures-MRD55 (other names: Mental retardation, autosomal dominant 55, with seizures) with intellectual developmental disorder autosomal dominant 55 | |
| 3-Methylglutaconic Aciduria, III-MGCA3 Type (other names: MGA III-MGA3 type, Optic atrophy plus syndrome, Optic atrophy with dystonia and spastic paraplegia, Iraqi-Jewish optic atrophy plus, Costeff syndrome, Optic atrophy-3 autosomal recessive) | OPA3 |
| Optic Atrophy Type 3 with Cataract (other names: Optic atrophy 3 autosomal dominant-OPA3, Optic atrophy and cataract autosomal dominant) | |
| Ornithine Transcarbamylase (OTC) Deficiency (other names: Hyperammonemia due to ornithine transcarbamylase deficiency, Ornithine carbamoyltransferase deficiency, OTC deficiency) | OTC |
| Phenylalanine Hydroxylase Deficiency (other names: Phenylketonuria, PAH deficiency, Phenylpyruvate deficiency, Folling's disease, Hyperphenylalaninemia, Non-PKU mild) | PAH |
| Hyperphenylalaninemia, BH4 Deficiency, D-HPAH4D (other names: Hyperphenylalaninemia due to tetrahydrobiopterin deficiency caused by Pterin-4α-carbinolamine dehydratase deficiency, Hyperphenylalaninemia with primapterinuria, CADH deficiency, PCBD deficiency) | PCBD1 |
| Peroxisome Biogenesis Disorder 1A (Zellweger) (other names: ZS - ZWS, Cerebrohepatorenal syndrome-CHR) | PEX1 |
| Peroxisome Biogenesis Disorder 1B (NALD/IRD) (other names: Peroxisome biogenesis disorder (neonatal adrenoleukodystrophy/infantile Refsum disease), Peroxisome biogenesis disorder (NALD/IRD), Adrenoleukodystrophy neonatal autosomal, Refsum disease, infantile, Phytanic acid storage disease) | |
| Heimler Syndrome 1 (other names: Sensorineural hearing loss with enamel hypoplasia, Nail defects peroxisome biogenesis disorder 1C-PBD1C) | |
| Peroxisome Biogenesis Disorder 6A (Zellweger) | PEX10 |
| Peroxisome Biogenesis Disorder 6B | |
| Peroxisome Biogenesis Disorder 14B | PEX11B |
| Peroxisome Biogenesis Disorder 3A (Zellweger) | PEX12 |
| Peroxisome Biogenesis Disorder 3B | |
| Peroxisome Biogenesis Disorder 11A (Zellweger) | PEX13 |
| Peroxisome Biogenesis Disorder 11B | PEX13 |
| Peroxisome Biogenesis Disorder 13A (Zellweger) | PEX14 |
| Peroxisome Biogenesis Disorder 8A (Zellweger) | PEX16 |
| Peroxisome Biogenesis Disorder 8B | |
| Peroxisome Biogenesis Disorder 12A (Zellweger) | PEX19 |
| Peroxisome Biogenesis Disorder 5A (Zellweger) | PEX2 |
| Peroxisome Biogenesis Disorder 5B | |
| Peroxisome Biogenesis Disorder 7A (Zellweger) | PEX26 |
| Peroxisome Biogenesis Disorder 7B | |
| Peroxisome Biogenesis Disorder 10A (Zellweger) | PEX3 |
| Peroxisome Biogenesis Disorder 10B | |
| Peroxisome Biogenesis Disorder 2A (Zellweger) | PEX5 |
| Peroxisome Biogenesis Disorder 2B | |
| Achondrogenesis Type 5 | |
| Heimler Syndrome 2 (other name: Peroxisome biogenesis disorder 4C-PBD4C) | PEX6 |
| Peroxisome Biogenesis Disorder 4A (Zellweger) | |
| Peroxisome Biogenesis Disorder 4B | |
| Peroxisome Biogenesis Disorder 9B (other name: Refsum Disease Adult 2, Peroxisome biogenesis disorder Pex7-related atypical) | PEX7 |
| Chondrodysplasia Punctata Type 1 (other names: Peroxisome biogenesis disorder 9-pbd9, Punctate Rhizomelic Form-CDPR, Calificans Punctata) | |
| Glycogen Storage Disease VII (other names: GSD VII, Muscle phosphofructokinase deficiency, PFKM deficiency, Tarui disease) | PFKM |
| Glycogen Storage Disease X (other names: GSD X, Phosphoglycerate mutase deficiency myopathy due to phosphoglycerate mutase muscle deficiency, PGAMM deficiency) | PGAM2 |
| Congenital Disorder of Glycosylation, Type I-CDG1T (other names: CDG It, Phosphoglucomutase 1 deficiency, PGM1 deficiency, Glycogen storage disease XIV-GSD13, GSD XIV) | PGM1 |
| Glycogen Storage Disease IIXd (other names: GSD IXd, Muscle phosphorylase kinase deficiency, Muscle glycogenosis X-linked) | PHKA1 |
| Glycogen Storage Disease IIXa (other names: Liver glycogenosis X-linked type I-XLG1 type, Glycogen storage disease VIII, previously GSD8) | PHKA2 |
| Glycogen Storage Disease IIXb (other names: GSD IXb, Autosomal recessive glycogenosis of liver and muscle, Hepatic and muscle phosphorylase kinase deficiency, Autosomal recessive) | PHKB |
| Glycogen Storage Disease IXc-GSD IXc | PHKG2 |
| Refsum Disease (other names: Refsum Disease Adult 1, Phytanic Acid Oxidase Deficiency, Heredopathia Atactica Polyneuritiformis, Hereditary motor and sensory neuropathy IV-HMSN4, HMSN IV) | PHYH |
| Congenital Disorder of Glycosylation, Type Ia-CDG Ia type (other names: Jaeken syndrome, Phosphomannomutase 2 deficiency, CDG type Ia) | PMM2 |
| Maple Syrup Urine Disease, Mild Variant | PPM1K |
| Neuronal Ceroid Lipofuscinosis 1 (other name: Ceroid lipofuscinosis 1 late infantile) | PPT1 |
| Cardiomyopathy, Fatal Congenital Glycogenosis of the Heart (other names: Phosphorylase kinase heart deficiency, Glycogen storage heart disease) | PRKAG2 |
| SAP Complex Deficiency Syndrome (other names: Prosaposin deficiency syndrome, Saposin complex deficiency syndrome) | PSAP |
| Gaucher Disease, Atypical (other name: Saposin C deficiency) | |
| Krabbe Disease, Atypical (other name: Saposin A deficiency) | |
| Variant White Matter Disease due to SAP-B Deficiency (other names: Leukodystrophy, metachromatic due to cerebrosidase activator deficiency, Saposin B deficiency) | |
| Hyperphenylalaninemia, BH4 Deficiency, A (other names: Hyperphenylalaninemia due to PTS deficiency, Tetrahydrobiopterin deficiency due to 6-pyruvoyltetrahydropterin synthase deficiency, PTSD, PTS deficiency) | PTS |
| Glycogen Storage Disease VI | PYGL |
| McArdle Disease (other name: Myophosphorylase deficiency) | PYGM |
| Hyperphenylalaninemia, BH4 Deficiency, C (other names: Hyperphenylalaninemia due to DHPR deficiency, Tetrahydrobiopterin deficiency due to dihydropteridine reductase deficiency, DHPR deficiency, QDPR deficiency) | QDPR |
| Congenital Disorder of Glycosylation, Type In | RFT1 |
| White Matter Brain Disease with Dystonia and Motor Neuropathy (other name: Sterol carrier protein 2 deficiency) | SCP2 |
| Hearing Loss, Encephalopathy, Leigh-Like Syndrome with 3-Methylglutaconic Aciduria Type VI-MGCA6 | SERAC1 |
| Mucopolysaccharidosis IIIA (Sanfilippo A) (other names: MPS IIIA, Sanfilippo Syndrome A, Heparan sulfate sulfatase deficiency, SGSH deficiency) | SGSH |
| Hyperinsulinemic Hypoglycemia, Familial, 7-HHF7 (other name: Exercise-induced hyperinsulinemic hypoglycemia) | SLC16A1 |
| Sialic Acid Accumulation Infant (other names: Sialuria infantile, N-acetylneuraminic acid storage disease, NSD) | SLC17A5 |
| Salla Disease (other name: Sialuria Finnish Type) | |
| Systemic Primary Carnitine Deficiency (other names: Systemic carnitine deficiency - SCD, Systemic carnitine deficiency due to renal reabsorption disorder, Primary carnitine deficiency, Carnitine transporter plasma membrane deficiency, Carnitine uptake defect - CUD) | SLC22A5 |
| Citrullinemia, Adult-onset Type II (other name: Citrin deficiency) | SLC25A13 |
| Citrullinemia Type II, Neonatal-onset (other name: Citrullinemia type II, neonatal onset, presence or absence of sibutramine and lipid abnormalities, neonatal intrahepatic cholestasis due to citrin deficiency - NICCD) | |
| Hyperammonemia-Hyperornithinemia-Homocitrullinuria (HHH) Syndrome (other name: HHH syndrome, ornithine translocase deficiency) | SLC25A15 |
| Carnitine-Acylcarnitine Translocase (CACT) deficiency (other name: CACT deficiency) | SLC25A20 |
| Fanconi-Bickel Syndrome-FBS (other names: Hepato-renal glycogenosis with renal falconi syndrome, Hepatic glycogenosis with Fanconi syndrome, Amino aciduria and glycosuria with hepatic glycogenosis, Fanconi syndrome with intestinal absorption and galactose intolerance, Pseudo-phloridzin glycosuria, Glycogenosis type XI) | SLC2A2 |
| Congenital Disorder of Glycosylation IfCDG IF Type | SLC35A1 |
| Congenital Disorder of Glycosylation IIm Type (other names: CDG IIm, developmental and epileptic encephalopathy 22-DEE22, early infantile epileptic encephalopathy 22-EIEE22) | SLC35A2 |
| Congenital Disorder of Glycosylation IIc-CDG IIc Type (other names: leukocyte adhesion deficiency II-LAD2 type, Rambam-Hasharon syndrome-RHS) | SLC35C1 |
| Congenital Disorder of Glycosylation IIw Type | SLC37A4 |
| Glycogen Storage Disease IIb - GSD IIb (other name: Glucose-6-phosphate transport defect) | |
| Glycogen Storage Disease Ic - GSD Ic | |
| Congenital Disorder of Glycosylation IInCDG IIn tType | SLC39A8 |
| Cerebral Creatine Deficiency Syndrome 1-CCDS1 (other names: X-linked creatine deficiency syndrome, creatine transporter deficiency, seizures associated with mental retardation X, low stature and midface hypoplasia, mental retardation X associated with creatine transport deficiency) | SLC6A8 |
| Hyperglycinemia without Normal Serum gGlycine | SLC6A9 |
| Niemann-Pick Disease Type A (other names: Sphingomyelin lipidosis, Sphingomyelinase deficiency, Acid sphingomyelinase deficiency neurovisceral type, ASMD neurovisceral type) | SMPD1 |
| Niemann-Pick Disease Type B (acid sphingomyelinase deficiency visceral type ASMD visceral type) | |
| Congenital Disorder of Glycosylation Iq Type (other names: colon tumors with brain malformations and endocrine abnormalities in ichthyosis, CDG Iq) | SRD5A3 |
| Caroli Syndrome (other names: mental retardation cataract, colonic tumor, and spinal lordosis autosomal recessive) | SRD5A3 |
| Congenital Disorder of Glycosylation IyCDG IY type | SSR4 |
| Congenital Disorder of Glycosylation Iw type | STT3A |
| Congenital Disorder of Glycosylation Iw Type | |
| Congenital Disorder of Glycosylation Ix-CDG Ix Type | STT3B |
| Mitochondrial DNA Depletion Syndrome 5 (other names: Mitochondrial DNA depletion syndrome, brain-muscle type with or without methylmalonic aciduria, autosomal recessive SUCLA2-related) | SUCLA2 |
| Mitochondrial DNA Depletion Syndrome 9 (other name: Lactic acidosis, previously lethal) | SUCLG1 |
| Multiple Sulfatase Deficiency (other names: Mucosulfatidosis, Sulfatidosis, Juvenile Austin type) | SUMF1 |
| Barth Syndrome (other names: mental retardation cataract, colonic tumor, and spinal lordosis autosomal recessive) | TAFAZZIN |
| 3-Methylglutaconic Aciduria Type IX (MGCA9) | TIMM50 |
| Congenital Disorder of Glycosylation Ik-CDK Ik Type | TMEM165 |
| Congenital Disorder of Glycosylation IpCDK Ip Type | TMEM199 |
| Mitochondrial Complex V (ATP Synthase) Deficiency, Nuclear Type 2 (MC5DN2) (other name: Mitochondrial newborn with TMEM70-type brain myocardial disease due to ATP synthase deficiency) | TMEM70 |
| Neuronal Ceroid Lipofuscinosis 2 (CLN2) (other names: Ceroid lipofuscinosis neuronal 2 variable age, onset, Jansky-Bielschowsky disease) | TPP1 |
| Spinal Cerebellar Ataxia, Autosomal Recessive 7 | |
| Intellectual Disability, Autosomal Recessive 7-MRT7 (other names: Intellectual disability 22-MRT22, Mental retardation autosomal recessive 7-MRT7, Mental retardation autosomal recessive 22) | TUSC3 |
| Mucopolysaccharidosis Plus Syndrome | VPS33A |
Kits
| Carrier Screening Core kit |
| Analyzes 19 genes of individuals with unknown carrier status. |
| Carrier Screening Comprehensive kit |
| Analyzes 228 genes of individuals with unknown carrier status. |
| PGT kit |
| Enables detection of all chromosome aneuploidies, structural abnormalities up to 10Mb, several types of Y chromosome aneuploidies, and mosaicisms exceeding 50%. |
| Hereditary Cancer Kit |
| Analyzes 62 genes covering 24 cancer predisposition syndromes associated with hereditary tumors. |
| Infertility Kit |
| Analyses 54 genes in the female infertility panel, 39 genes in the male infertility panel, including both structural and numerical abnormalities of sex chromosomes. |
| Neonatal kit |
| Analyses 140 genes for symptomatic and pre-symptomatic infants. |
| Cardiac Comprehensive Kit |
| Analyses 292 genes covering major hereditary cardiovascular diseases. |
| Cardiomyopathy Kit |
| Analyses 98 genes covering hereditary cardiovascular diseases associated with cardiomyopathy. |
| Arrhythmia Kit |
| Analyses 42 genes covering hereditary cardiovascular disorders related to arrhythmias. |
| Aortopathy |
| Analyses 48 genes covering hereditary cardiovascular diseases associated with aortopathy. |
| Congenital Heart Defects Kit |
| Analyses 80 genes covering congenital heart defects. |
| FH, PH, RAS |
| Analyzes 11 genes for Familial Hypercholesterolemia (FH), 11 genes for Pulmonary Hypertension (PH), and 30 genes for RASopathies (RAS). |
| Metabolic Kit |
| Analyses 223 genes covering major classes of hereditary metabolic disorders. |
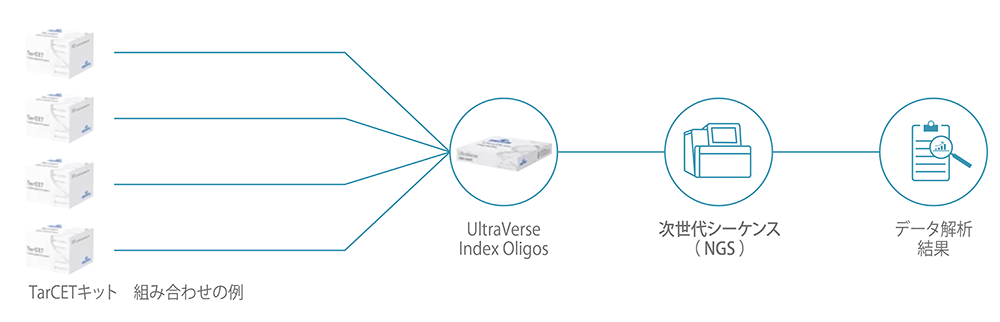
| UltraVerse Index Oligos Type A/B/C/D |
| Contains proprietary dual index oligos compatible with all test kits. |
Each test kit contains reagents for 16 reactions with a maximum of 384 multiplexing capabilities (4 x 96 index kits).
Each UltraVerse Index Oligos kit contains 96 reactions.
Proprietary Software Solutions
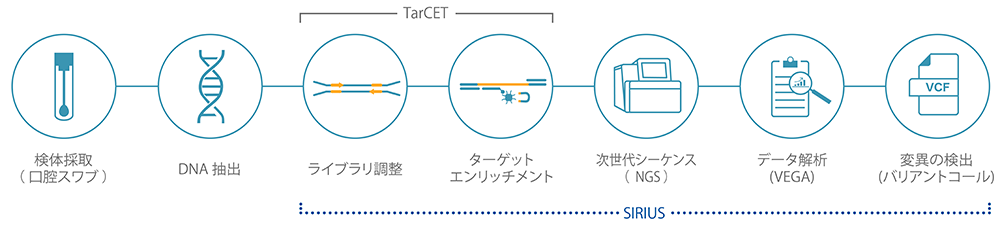
Workflow
SIRIUS

SIRIUS is a data management web application that enables user management of information generated by NGS analysis when processing samples using the workflow of the test kit. Additionally, SIRIUS facilitates the creation, computation, and editing of sample batches required for sequencing and collaborates with Medicover Genetics' analysis engine, VEGA, to provide information about analysis data in visual graphs and tabular formats.
VEGA

VEGA is a bioinformatics analysis software for samples processed using the test kit. VEGA analyzes NGS data generated by the genetic testing workflow. This analysis detects single nucleotide variants, short insertions or deletions, and changes in copy number. In PGT analysis, aneuploidy and structural abnormalities are detected.
Support

Medicover Genetics provides continuous support to all customers of TarCET kits through dedicated technical support channels. We are able to offer technical support for troubleshooting, quality, and performance monitoring at any time. Upon request, we provide training at certified facilities to our customers on workflows, protocols, and data analysis.
For inquiries regarding technical support:
info@humedit.com


